|
|
|
Huntington’s disease (HD) is a devastating genetic disorder that robs its victims of physical and mental control. Resulting from the mutated HTT (Huntington’s) gene, it is one of the most destructive diseases known to man. There are approximately 30,000 symptomatic patients in the US and more than 200,000 at-risk of inheriting HD, usually diagnosed between the ages of 30 and 45. It impairs the patient’s ability to lead a normal life due to progressive chorea (abnormal involuntary movements) accompanied by cognitive and psychiatric problems. No treatment is available that can stop or reverse the course of HD and the average life expectancy following diagnosis is only about 10-30 years.
The cause of HD symptoms is monogenic, resulting from the accumulation of toxic protein aggregates causes by the mutant HTT1 gene. One potential solution is to add the wild type Huntington protein; however, this must be provided for the patient’s lifetime and the protein is expressed and required in many different cells. There is a dire need for efficacious, safe therapies that decrease symptoms and/or modify the disease.
Few drugs have been approved for treating HD patients, although there has been global growth in HD therapeutic research. Some of the major companies involved are Valeant, Lundbeck, Prana Biotech, Teva, SOM Biotech, GlaxoSmithKline, Siena Biotech, Raptor, Pfizer, and Ipsen.
|
|
|
Therapies in Development
One in every 10,000 patients has difficulty in eating, drinking, communicating, and exhibits psychiatric symptoms. As in many other neurological diseases, symptomatic treatment is the only option and does improve the quality of life and prevent complications. Xenazine was the first US drug approved (2008) and recently, the HD community was encouraged by the FDA’s approval of Teva’s “Austedo” (February 2017), the first in 9 years. Both drugs are used for the treatment of chorea associated with HD. Other drugs prescribed “off-label” include haloperidol and olanzapine for motor symptoms, donepezil, rivastigmine, and riluzole for cognitive symptoms, and citalopram and risperidone for psychiatric symptoms. However, the long-term use of these drugs has a range of side-effects and is not suitable for all HD patients.
In spite of considerable attention from scientists since the early 20th century, there is still a strong unmet medical need with most new, disease-modifying assets in the preclinical stage. According to clinical trial intelligence, approximately 140 trials have been conducted with 80 completed. Of these, 46 were Phase I or II studies and of these 8 were terminated and 1 suspended (the status 20 was unreported).
|
|
|
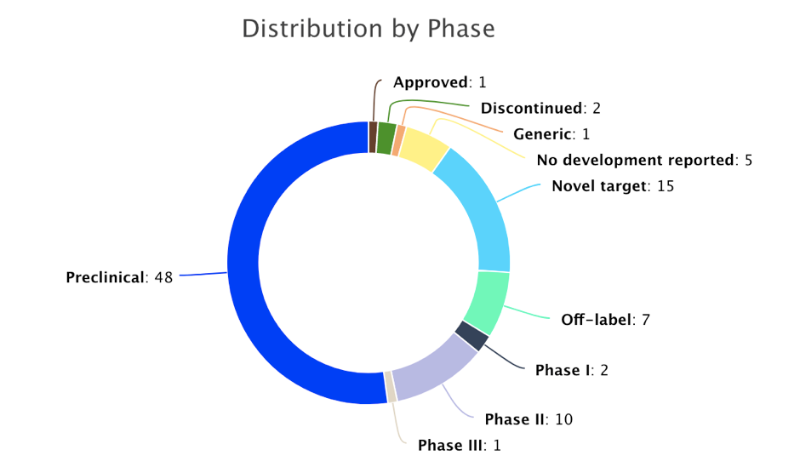
PharmGPS® discloses above 37 active trials (Phases I, II, III, approved and novel targets), of which 9 are completed and 11 ongoing. PharmGPS® also provides the assessment of 14 pipeline drugs in clinical development for HD. Antioxidant “cysteamine delayed release” is the only Phase III drug. PBT2 and Huntexil (both in Phase II) have demonstrated efficacy and safety. A novel modality drug, gene silencing, is in clinical trials (Ionis Pharmaceuticals (IONIS-HTT Rx) and has received orphan drug designation (EMA & FDA). New clinical entrants which have not yet been evaluated include PBF-999, ANX00, and ORY-2001. In addition, there are 15 novel targets (gene therapy, stem cell therapies) in preclinical.
|
|
|
The PharmGPS®Analysis
Huntexil®: Huntexil® is an oral dopamine D2 stabilizer that improves HD movement symptoms and has shown significant Phase III efficacy on the modified motor score (mMS). These results were observed without any of the usual side effects, such as sedation and depression. Based on clinical trial evidence, Teva believes that Huntexil® holds promise for symptomatic relief and will increase the quality of life for patients.
Cysteamine Delayed Release: RP103 is a cystine-depleting agent which has shown 23% slowing in the rate of decline in total functional capacity and 46% slowing in the rate of deterioration (Independence Scale, primary endpoints) and favoring early treatment intervention in a Phase II/III trial. Unfortunately, these results were not significant, although treatment was safe and well tolerated.
PBT2: PBT2 is a neuroprotective agent that reduces the interaction between the HTT protein and copper in the brain, inhibiting several harmful effects of copper. In Phase II, it has shown significant safety and tolerability (primary endpoints, Reach2HD trial).
PharmGPS® analysis predicts that Huntexil could be a successful drug for HD based on Phase III evidence with improvement in hand movements, gait, and balance. Researchers are also developing treatments that reduce toxic protein aggregation by turning down the expression of aberrantly expressed genes.
The Huntington’s Disease Market Will Show Significant Growth Over the Next Decade
HD is a highly unaddressed indication with ~60,000 patients across the major global markets and only two approved drugs: Xenazine (2008) and Austedo (2017). The current market for the US, EU, and Japan is ~$400 M and is expected to grow at a CAGR of 25.3% to reach $3.0 B by 2026. Market growth is likely to be driven by the launch of disease modifying drugs, such as Raptor’s RP103 (cysteamine delayed release), Teva’s Huntexil, and Prana’s PBT2, to address the underlying, high unmet need. The market is expected to be dominated by the US, accounting for approximately 75% of future HD drug sales globally.
|
|
|
| Download the PharmGPS GO App |
|
 |
 |
|
|
|
| |
| |
|
| |
|
|